When it comes to understanding cancer, getting our heads around the complicated process of how it forms in the first place is vital. When we know what drives cancer then we can figure out how to diagnose it and ultimately, how to treat it.
So far, research has shown us that the vast majority of cancers are caused by some level of genetic mutation. This is something we’ve known for a long time and something we use in order to treat different types of cancer. For example, a reasonable proportion of breast cancer patients have a mutation in the gene for a particular protein called HER2 making this protein more abundant than it should be and allowing cancer cells to grow and grow. So scientists have designed a therapy that blocks, or inhibits, HER2 – a drug called Herceptin. The cancer cells have been relying on HER2 for their growth, they’ve becomes entirely dependent on it which means when we treat patients with Herceptin, HER2 activity is reduced and the cancer cells die.
Another thing we know about cancer, is that it’s hugely genetically unstable. This means cancer cells acquire loads and loads (sometimes many hundreds) of new mutations as the disease progresses. What is important is to understand which of these many mutations are actually fundamental to the survival of the cancer. Which ones are truly driving the disease, allowing it to survive and progress and spread?
This can be particularly complicated for us to understand because a mutation in one gene can do lots of different things.
Genes
First things first, let’s define what a gene is. When it comes to DNA we can think of an organism’s entire genome like a recipe book. The entire human genome book allows our cells to work together to cook an entire human. If we were cooking a really complicated meal like Christmas dinner we would need to cook all the different components and bring them all together to make the meal. You have to cook the main dish and all the side dishes, your carrots and parsnips and brussel sprouts, maybe two kinds of potatoes and a soup for the starter. Every person’s Christmas dinner might look slightly different but we all agree it’s a complicated meal and there are lots of components. The human body is also pretty complicated and is made up of components. A human body is made up of organs, which at the most basic level are made up of cells. Your cells are kind of like the ingredients for each dish in your Christmas dinner. Your organs are kind of like those individual dishes. They all come together to make the whole human. So, in order to make such a complicated thing, we need lots and lots of recipes. Within the human genome we have 23 pairs of chromosomes, which we might think of as chapters of the recipe book. Within each chapter are individual recipes, or genes, which allow cells to make for individual building blocks (ok, ok, I’m mixing my metaphors…).
The basic building blocks in the human body are cells. Cells come together to make our organs, and organs come together to make a human. But our cells need to do specific jobs depending on what kind of cell they are; they need to communicate with one another within the organs they are a part of. This is what makes our heart cells contract all together to pump blood around the body or it allows all of our lung cells to know that they’re supposed to be lung cells. This communication is done through proteins, which can pass signals along complicated pathways transmitting the message from protein to protein and giving each cell instructions on how to function. A single gene is a recipe for an individual protein. Each gene is made up of many thousands (depending on the protein size) of what we call bases which are signified by the letters A, T, C and G. There is an extra layer of complexity as the code that makes up a single gene is organised into groups of three letters each group referred to as a codon. Each codon codes for a single amino acid and these amino acids build up in long sequences to eventually form proteins.

A mutation is change in this code. And that tiny change can have a number of outcomes.
Ultimately – the ones we’re worried about are those that change how particular proteins behave.
How do protein mutations drive cancer?
On a very basic level, proteins basically control everything. They regulate which cells go where, how big cells grow, how much cells replicate and how cells function within their organs. Some proteins are crucial to very specific pathways related to cell survival and cell replication (referred to as proliferation). These proteins we refer to as oncogenes or tumour suppressors. They carefully regulate how cells grow and survive. If these pathways become deregulated, cells can grow out of control and survive when they shouldn’t. This is how cancer forms. These pathways, and therefore these particular proteins, are fundamental in driving cancer. Many of the most commonly mutated proteins in cancer are either oncogenes or tumour suppressors. But what exactly are these proteins?
An oncogene is a protein which promotes cell growth and cell survival. In normal cells these proteins are carefully regulated. They become activated and they pass on the message to tell the cell to grow or survive and then they get switched off until they are next required. Oncogenes can be mutated in such a way that their activity is increased – either there are loads of copies of them all passing on the “grow and survive” message at once, or they are unable to be switched off and they are constantly telling the cell to grow and survive. In this way, a mutation in an oncogene can drive cancer.
A tumour suppressor is one of those proteins that helps regulate the growth and survival pathways. Tumour suppressors can switch oncogenes off, or they can oppose them by sending the opposite message. They serve to slow down the “grow and survive” message and keep our cell growth and survival finely tuned. In this way, when functioning properly, tumour suppressors prevent cancer. A mutation that blocks these proteins from doing their job will therefore promote cancer.
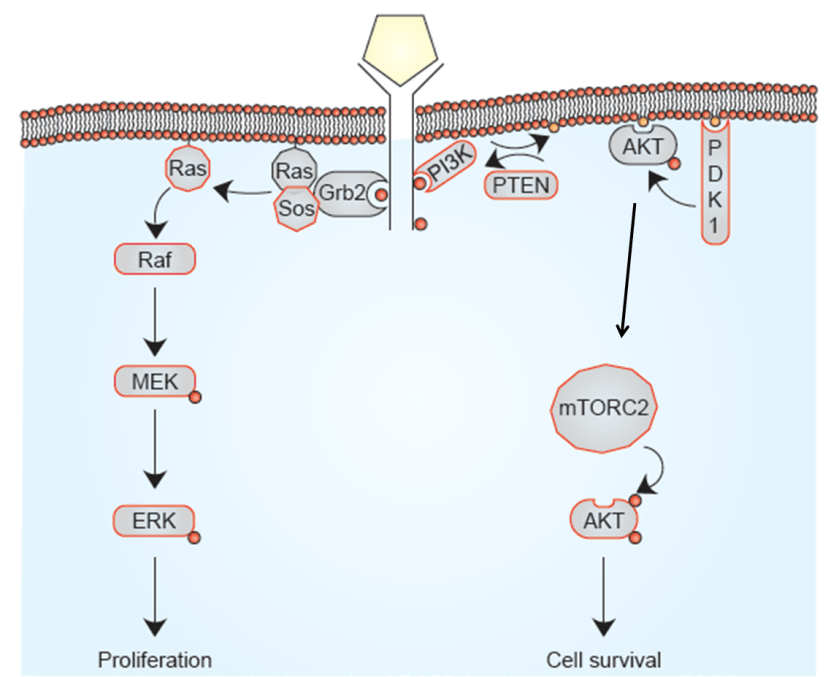
Different types of mutation
This complexity of the genetic code means there are number of different ways in which a gene can be mutated.
We can split these into two broad groups – synonymous and non-synonymous. Synonymous mutations are ones where the mutation doesn’t lead to change in the sequence of amino acids in a particular protein. They are effectively silent mutations because they don’t change proteins.
But many gene mutations are non-synonymous; one way or another, they cause a change in the protein produced by that gene.
Of these we can have:
Missense mutation is a mutation where one amino acid is switched out for another. The importance of that particular amino acid to that particular protein designates whether or not this change has a big impact on the protein function. It can increase activity of an oncogene, or it can prevent an active oncogene from being switched off. Alternatively, it can block the action of a tumour suppressor.
Nonsense mutation is a mutation which leads to termination of the protein production at that position in the code, leading to a shortened or truncated version of the protein which may severely alter the ability of that protein to function normally. A truncated tumour suppressor, for example, isn’t able to switch off cell growth and might promote cancer.
Insertions/deletions occur when a few letters are added to or removed from the code which can completely alter the entire remaining sequence leading again to protein truncation. Large deletions can remove entire genes preventing the production of that entire protein. This can include duplication mutations when a particular region of DNA is duplicated which may enhance or reduce the activity of a protein.
Large-scale mutations in chromosomal structure – these can be very complicated including the amplification of entire genes giving you far too many copies of a particular protein. They can also create entirely new proteins by fusing two separate genes together.
What does all this mean?
Hopefully it’s clear that the variety of mutations and proteins involved in driving cancers is quite complicated and that makes understanding cancer difficult. However, we might also see opportunities for using knowledge to treat cancers. If we can establish how frequently a particular cancer is cause by an overactive oncogene then we can start to find treatments that block or inhibit those oncogenes. These are strategies that have led us to discover drugs like Herceptin, which blocks the oncogenic HER2 receptor I talked about at the beginning of this post. But it also means we need to fully understand which mutations drive cancer in order to establish treatments for them.
Next week I’m going to talk about a particular study that looked at these driver mutations that was published just this month. The researchers wanted to get a more complete picture of how many proteins we might need to target in order to effectively treat specific types of cancer and what they found was really interesting.
For now, I’d love some feedback on this post because it’s quite science heavy – if there’s anything I could have explained better or in more detail then let me know in the comments and maybe I can write more on these topics in the future.
*edited to correct some of the HER2 science – thanks to my good friend Dr Jenny Smith-Wymant for pointing out my mistake.


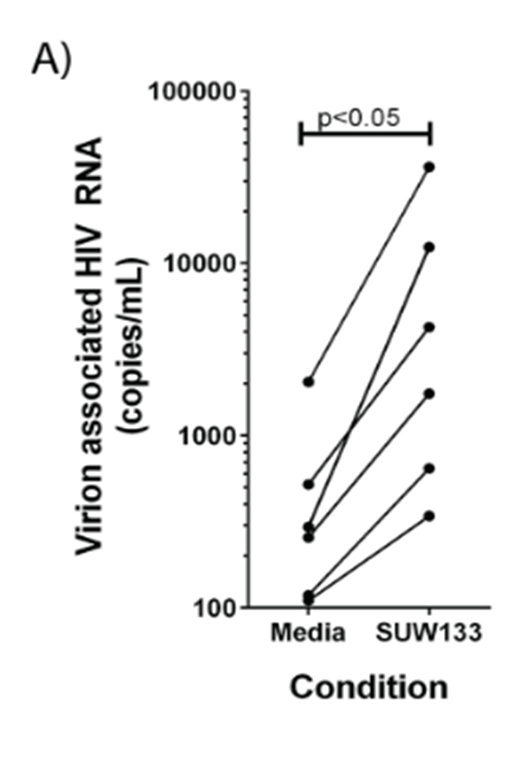
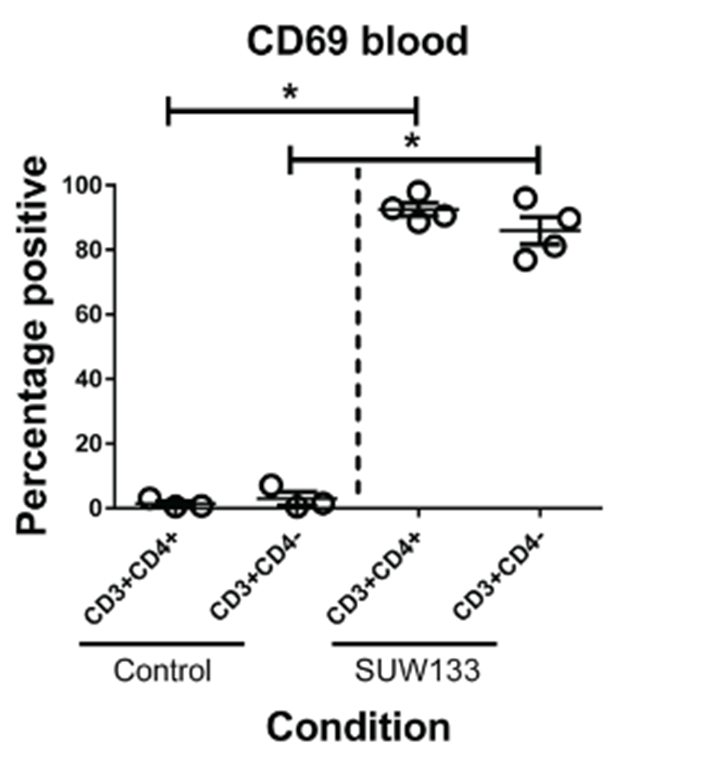




 Image: figure from the
Image: figure from the