So far on this blog, I’ve talked a lot about cancer. I’ve been working in cancer cell biology for a number of years, and it’s an area that a lot of people are interested in, so it makes for a sensible topic. By training, though, I am a cellular and molecular physiologist. I’m interested in how all cells work; they don’t have to be cancer cells.
The human body, and cells in particular, amaze me. I love learning the intricate details of exactly how our cells work on a molecular level, but I love how we can identify little changes that might be useful in understanding disease and treatment even more.
Earlier this year I came across an interesting study on treating individuals infected with the Human Immunodeficiency Virus (HIV). HIV is a complicated virus and has long been a source of fear. But by understanding the science of the virus we can look to treat infection and perhaps dispel some of those fears. So here’s a bit about HIV, and then a bit about a cool study.
HIV
HIV is a type of virus that we call a retrovirus. A retrovirus carries its genetic material as a single strand of RNA, instead of the double-strand of DNA we use as humans. To transmit its genetic material the virus needs to construct DNA, which it does inside a host cell. In the case of HIV, the host cells are cells within the human immune system – white blood cells called helper T cells. Once an HIV molecule (a virion) gets inside a human host cell, it uses an enzyme within the virus to transcribe a piece of its RNA into DNA. It then slots that DNA into the host cell genome. It can remain dormant for many years but eventually the host cell machinery produces many, many copies of this piece of DNA, releasing more HIV virions into the blood stream. Essentially, the virus hijacks the host cell’s normal processes in order to make copies of itself.

The HIV virus does not need all the machinery required for its own replication therefore the HIV genome is tiny, consisting of only nine genes. By comparison, the human genome consists of around 20,000 genes!
Treating HIV
These days we are pretty good at treating and managing HIV infection. HIV infection isn’t currently curable, but we’re so good at treating it that many patients live a long, healthy life while carrying HIV. There are many caveats to that, including political problems related to getting treatment to all the parts of the world that need it.
The very first treatment we produced for HIV was an inhibitor that blocked the action of a protein important in converting the virus’ RNA into DNA, so preventing its subsequent replication and spread throughout the body. Since then we’ve designed different types of antiretroviral therapy (ART) which target different processes within the virus’ lifecycle. We can stop the virus getting into a cell, stop its RNA converting to DNA, stop that DNA integrating into the human genome and stop the virus from assembling and breaking out of the host cell. The problem is, once the virus is integrated into the host cell DNA it can lay dormant, untouched by inhibitors even when the viral load is reduced in the patients’ blood. We have to maintain treatment to prevent this dormant DNA from activating and the virus spreading around the human body. It is possible, too, for the virus become resistant to each therapy meaning careful assessment of the patient and modification of the therapy accordingly. In this way, we can manage the infection long-term. But what if we could figure out a way to target the dormant virus sitting in host cell DNA?
New study

A new study published in September aimed to do exactly this. Earlier research had shown that a particular type of drug can ‘activate’ the host-cells which hold dormant HIV DNA. These ‘activated’ cells produce lots and lots of RNA for the virus which “kicks” the hiding virus back into action. Using this principle, the standard anti-retroviral drugs are then able to “kill” the virus specifically targeting this pool of dormant virus that standard therapy alone is unable to target. This strategy is referred to as “kick and kill”. While other researchers have studied this principle before, the effect on host-cell activation has been minimal which means only a small proportion of cells holding dormant HIV can be targeted. The larger this proportion, the closer we can get to activating and killing every cell holding dormant-HIV and the closer we can get to curing HIV infection.
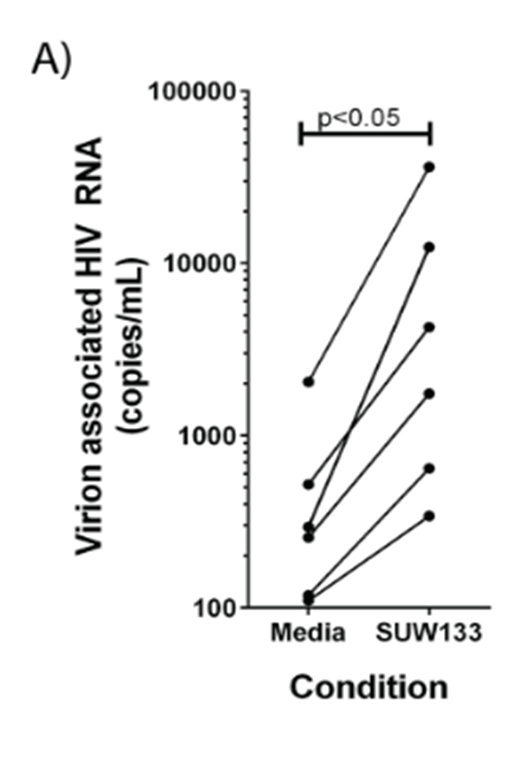
In this study the authors used a synthetic compound, SUW133, which would they hoped might activate more host cells and therefore be more successful. Their initial work looked at the effectiveness of the compound in cells taken from six patients. They showed that control treated cells (treated with only media) had relatively few RNA molecules from the HIV virus whereas when they took those same patient-derived cells and treated them with the SUW133 the number of viral RNA molecules increased significantly. This indicates that SUW133 does, indeed, activate the dormant virus and kicks the host cells into action.
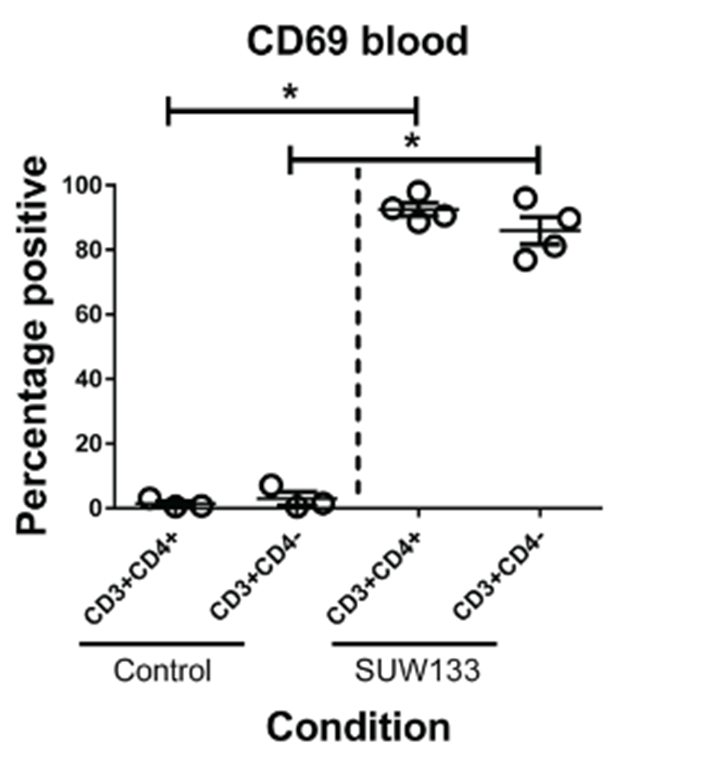
They then wanted to look at what would happen in a living organism, so the researchers had to infect humanised mice (mice genetically modified to express human genes) with HIV. They did this for 3-4 weeks, after which they treated the mice with antiretroviral therapy for 2-5 weeks. Once they were sure that only dormant HIV would remain in the mice, they treated them with their special synthetic compound and looked in the blood for evidence of HIV-host-cell activation. In mice treated with a control (inert) compound they found relatively low proportions of specific cell types (CD3+/CD4+ and CD3+/CD4-) that expressed a marker for activation (CD69) however upon treatment with the active compound (SUW133) the proportion of “activated” CD69 positive cells rose dramatically.
What’s more, the researchers showed that a small proportion (around 25%) of these activated cells were dead or dying which shows promise for the additional treatment with standard antiretroviral therapy to fully “kill” the virus.
Overall the authors have shown that they can activate dormant HIV which they hope will give us the opportunity to kill this currently untreatable pool of the virus and one day give us a cure to HIV infection.
What’s clear is that this is still early days – the researchers need to fully establish the effect of this “kick” strategy when combined with the “kill” treatment (ART). However, it is incredibly promising that we might someday be able to activate host-cells carrying dormant HIV genetic material so that we can remove those cells entirely. If we are successful, we could go from long-term HIV infection management to HIV-infection cure.


 Image: figure from the
Image: figure from the